
Multiple Endocrine Neoplasia (MEN) and other inherited endocrine syndromes are rare genetic conditions that increase the risk of developing endocrine tumors over a lifetime. These conditions may affect the thyroid, parathyroid, adrenal glands, pituitary gland, and neuroendocrine system.

At the Saint John’s Cancer Institute Genetic Counseling Program, patients receive a comprehensive and thoughtful evaluation that includes genetic testing, personalized risk assessment, and coordinated follow‑up care.
Our genetics and endocrine specialists work closely together to help you understand what an inherited endocrine syndrome may mean for you and your family. We guide you through each step of the process—from the initial consultation to long‑term surveillance—so you can make informed, confident decisions about your health.
When to Suspect an Inherited Endocrine Syndrome
Inherited endocrine syndromes are uncommon, but certain patterns raise concern for a genetic predisposition. Recognizing these signs early allows patients and families to receive appropriate testing and long‑term monitoring. Early recognition is essential, as timely genetic testing, surveillance, and treatment can significantly improve long‑term outcomes. Our endocrine and genetics teams work together to evaluate individuals who may be at risk.
Common Indicators
A genetic syndrome may be suspected when endocrine tumors occur at a young age, appear in more than one gland, or run in families. These features help guide decisions about genetic testing and surveillance.

- Medullary thyroid carcinoma
- Pheochromocytomas or paragangliomas
- Hyperparathyroidism diagnosed under age 30–40
- Functional pituitary tumors in younger patients
- More than one endocrine tumor in a single patient
- A family history of endocrine tumors
Why Early Evaluation Matters
A small proportion of endocrine tumors are inherited, and identifying a genetic cause can guide treatment, screening, and family counseling. Patients meeting these criteria are referred to our specialized genetics team for testing and long‑term risk management.
Multiple Endocrine Neoplasia Type 1 (MEN1)
Multiple Endocrine Neoplasia Type 1 (MEN1) is an inherited condition most often caused by mutations in the MEN1 gene. It increases the risk of developing tumors in the parathyroid glands, pituitary gland, and pancreas or gastrointestinal tract. Because MEN1 can affect several organs over time, lifelong monitoring is essential.
Key Features of MEN1
MEN1 commonly presents with primary hyperparathyroidism, pituitary tumors, and pancreatic neuroendocrine tumors. Additional tumors, including adrenal tumors or small bowel neuroendocrine tumors, may also occur.
- Primary hyperparathyroidism (nearly all patients)
- Pituitary tumors, often hormone‑secreting
- Pancreatic or gastrointestinal neuroendocrine tumors
- Occasional adrenal or small bowel neuroendocrine tumors
Diagnosis and Management
Diagnosis is confirmed through germline genetic testing and a detailed personal and family history. Patients with MEN1 require lifelong biochemical testing, imaging, and coordinated care. Surgical treatment may be recommended when tumors cause symptoms or hormonal changes.
Who Should Be Tested for MEN1?
Genetic testing is recommended for individuals with early‑onset hyperparathyroidism, four‑gland hyperplasia, or multiple MEN1‑associated tumors.
- Primary hyperparathyroidism under age 30–40
- Four‑gland parathyroid hyperplasia
- Two or more MEN1‑associated tumors
Multiple Endocrine Neoplasia Type 2 (MEN2)
Multiple Endocrine Neoplasia Type 2 (MEN2) is caused by mutations in the RET proto‑oncogene. MEN2 increases the risk of medullary thyroid cancer, pheochromocytoma, and, in some patients, hyperparathyroidism. Because certain RET mutations are associated with early or aggressive disease, genetic testing plays a critical role in management.
Core Features of MEN2
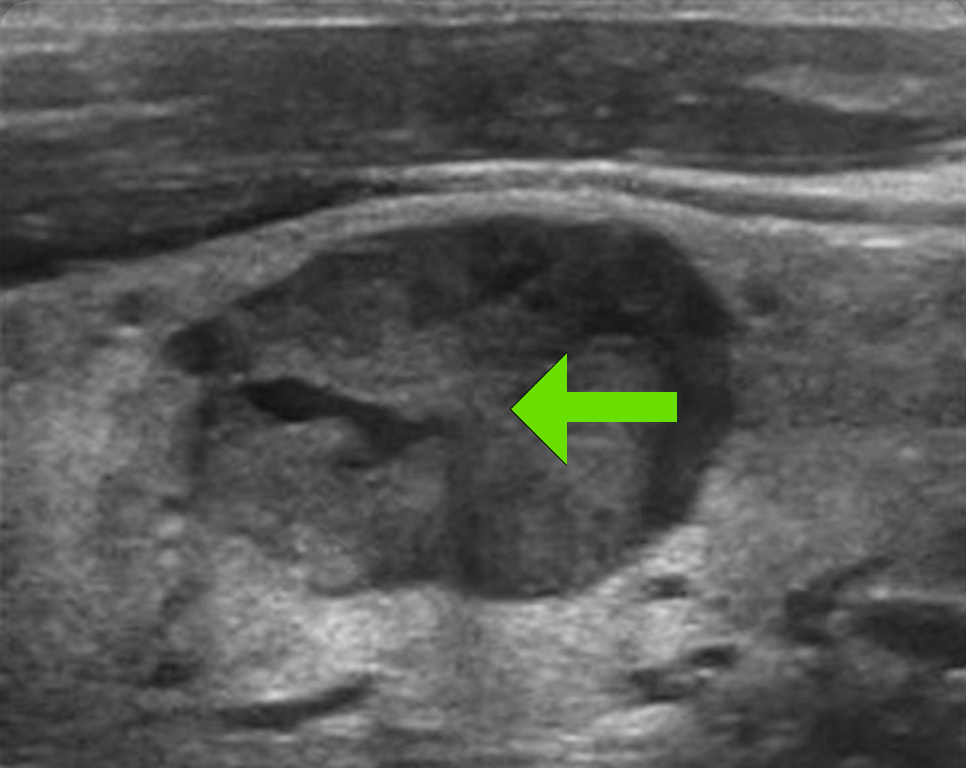
Medullary thyroid cancer is the hallmark of MEN2 and is present in nearly all affected individuals. Pheochromocytoma occurs in approximately half of patients, and some develop primary hyperparathyroidism.
- Medullary thyroid cancer
- Pheochromocytoma
- Primary hyperparathyroidism (in some patients)
- Mucosal neuromas or ganglioneuromas (depending on subtype)
MEN2 Subtypes
MEN2 includes MEN2A, MEN2B, and Familial Medullary Thyroid Cancer (FMTC). Each subtype has distinct features and risks based on the specific RET mutation.
MEN2A
MEN2A is characterized by medullary thyroid cancer, variable risk of pheochromocytoma, and occasional hyperparathyroidism.
MEN2B
MEN2B is the most aggressive subtype, with early‑onset medullary thyroid cancer, pheochromocytoma, mucosal neuromas, and a marfanoid body habitus.
Familial Medullary Thyroid Cancer (FMTC)
FMTC involves medullary thyroid cancer without other endocrine tumors. Patients require lifelong monitoring based on their specific RET mutation.
Hereditary Pheochromocytoma and Paraganglioma Syndromes
Nearly half of all pheochromocytomas and paragangliomas are associated with an inherited genetic mutation. These tumors may occur alone or as part of broader genetic syndromes. Identifying the underlying cause helps guide treatment and screening for patients and family members.
Common Associated Syndromes
Several inherited conditions increase the risk of pheochromocytoma or paraganglioma, including SDH‑related syndromes, Von Hippel–Lindau (VHL) syndrome, and neurofibromatosis.
- SDHA, SDHB, SDHC, SDHD mutations
- Von Hippel–Lindau (VHL) syndrome
- Neurofibromatosis type 1 (NF1)
- Neurofibromatosis type 2 (NF2)
Why Genetic Testing Is Recommended
Because these tumors may be the first sign of a broader inherited condition, all patients with pheochromocytoma or paraganglioma should be evaluated for genetic mutations. Early identification supports appropriate screening and family counseling.
Papillary Thyroid Cancer–Associated Syndromes
Although most papillary thyroid cancers are not inherited, certain genetic syndromes increase the risk. Identifying these conditions helps guide cancer screening, family evaluation, and long‑term management.
Associated Genetic Syndromes
Several inherited conditions are linked to an increased risk of papillary thyroid cancer.
- Familial adenomatous polyposis (APC)
- Li‑Fraumeni syndrome (TP53)
- Cowden syndrome (PTEN)
- Carney complex (PRKAR1A)
- DICER1 syndrome (DICER1)
Why Genetic Testing and Lifelong Surveillance Matter
Inherited endocrine syndromes often involve multiple tumors over time, sometimes beginning at a young age. Genetic testing helps identify at‑risk individuals, guide surveillance, and support early treatment. Ongoing monitoring is essential for reducing complications and improving long‑term outcomes.
Genetic testing for MEN is typically done with a simple blood test, but many laboratories also offer saliva‑based testing using a mouth swab. Both methods are accurate and used routinely for MEN1 and RET mutation analysis. Your genetics team will help determine which option is most appropriate.
Benefits of Genetic Testing
Testing helps identify family members who may be at risk, supports early detection of tumors, and guides personalized surveillance strategies.
- Identifying at‑risk relatives
- Early tumor detection
- Personalized screening plans
- Timely surgical or medical intervention
Patients with suspected or confirmed inherited endocrine syndromes are evaluated by a specialized team of endocrinologists, surgeons, and genetic counselors. Our coordinated approach ensures comprehensive care, from diagnosis and testing to long‑term surveillance and family support.

If you have questions regarding Multiple Endocrine Neoplasia or other endocrine-related conditions, please call today. Request an appointment



