
Specialized Care for Pheochromocytoma & Paraganglioma
Pheochromocytomas and paragangliomas (PPGLs) are rare neuroendocrine tumors that can cause the body to produce excess stress hormones, leading to serious symptoms such as high blood pressure, headaches, sweating, and heart palpitations. While uncommon, these tumors can have significant health consequences if left untreated.

At Providence Saint John’s Health Center, our Endocrine Tumor Program provides specialized, coordinated care for patients with adrenal and extra-adrenal neuroendocrine tumors.
We are focused on delivering personalized care that improves outcomes and supports long-term health.
What Are Pheochromocytoma & Paraganglioma?
Pheochromocytomas and paragangliomas are tumors that arise from chromaffin cells of the neuroendocrine system. These cells normally help regulate the body’s response to stress by producing catecholamines such as adrenaline and noradrenaline. When tumors develop, excess hormone release can occur—often in unpredictable episodes.
Pheochromocytoma
Pheochromocytomas develop in the adrenal glands, which sit above each kidney and play a key role in hormone regulation. These tumors are more likely to produce hormones and frequently cause sudden spikes in blood pressure and other symptoms.
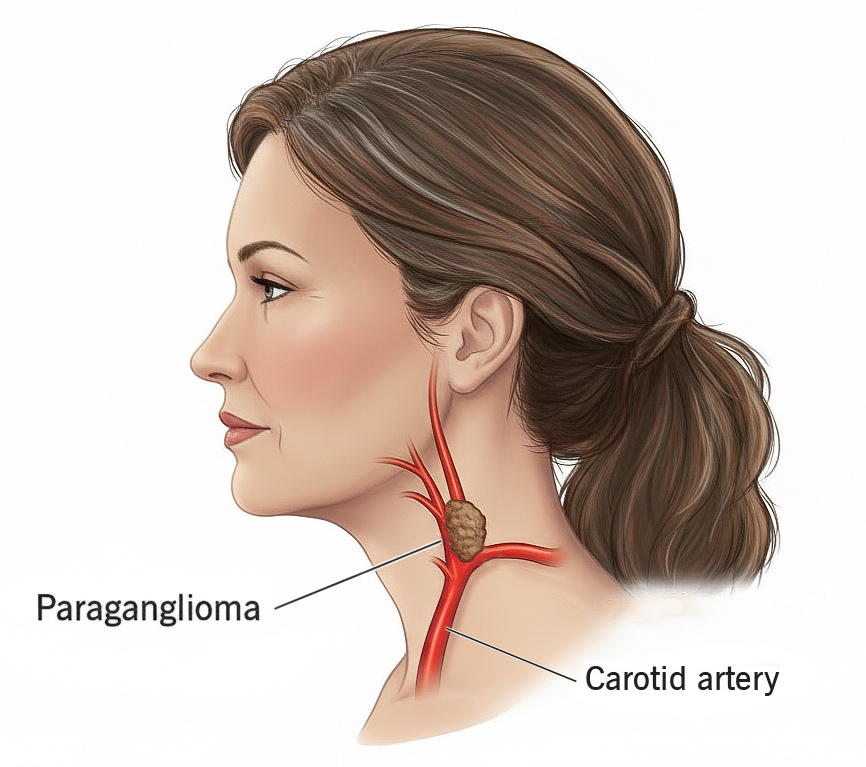
Paraganglioma
Paragangliomas form outside the adrenal glands, commonly along the spine or in the neck, chest, abdomen, or pelvis. Some paragangliomas produce hormones, while others do not. Because of their varied locations and behavior, diagnosis and treatment often require specialized imaging and multidisciplinary expertise.
Signs and Symptoms
Symptoms vary depending on whether the tumor produces hormones and may occur in episodes triggered by stress, physical activity, or certain foods.
Common symptoms include:
- High or episodic blood pressure
- Severe or recurring headaches
- Excessive sweating
- Rapid or irregular heartbeat
- Anxiety or panic-like episodes
Additional symptoms may include:
- Tremors
- Weight loss
- Heat intolerance
- Abdominal or chest pain (with larger tumors)
Some patients—especially those with nonfunctional tumors—may experience few or no symptoms. Even without symptoms, these tumors can grow and cause complications, underscoring the importance of early evaluation.
Causes and Risk Factors
Many pheochromocytomas and paragangliomas occur sporadically, with no known family history. However, up to 30–40% are linked to inherited genetic mutations.

Hereditary conditions associated with PPGLs include:
- Multiple Endocrine Neoplasia type 2 (MEN2)
- Von Hippel-Lindau (VHL) syndrome
- Hereditary paraganglioma-pheochromocytoma syndromes
- Neurofibromatosis type 1 (NF1)
Because of this strong genetic component, genetic counseling and testing are recommended for all patients diagnosed with PPGLs. Results help guide treatment decisions and long-term surveillance for patients and their families.
Diagnosis of Pheochromocytoma & Paraganglioma
Accurate diagnosis requires a step-by-step approach combining hormone testing, advanced imaging, and genetic evaluation.
Biochemical Testing
Initial testing determines whether excess hormones are present and may include:
- Plasma free metanephrines
- 24-hour urine metanephrines
- Catecholamine testing
These tests confirm whether the tumor is hormonally active.

Imaging Studies
Once hormone excess is identified, imaging is used to locate and characterize the tumor. Studies may include:
- CT or MRI of the abdomen and adrenal glands
- MRI of the chest, spine, or pelvis
- Functional imaging, such as MIBG scans or PET-CT for complex or metastatic disease
Genetic Testing
Given the high rate of hereditary involvement, genetic counseling is a critical part of care. Genetic findings influence treatment planning and determine appropriate long-term monitoring strategies.
Treatment Options
Treatment is tailored to each patient based on tumor location, hormone activity, genetic findings, and overall health. Care is coordinated across specialties to ensure safety and optimal outcomes.
Medical Preparation
Before treatment—especially surgery—patients often require medication to control blood pressure and heart rate, including:
- Alpha-blockers
- Beta-blockers
- Calcium-channel blockers

This preparation reduces the risk of dangerous hormone surges and supports safer treatment.
Surgery (Primary Treatment)
Surgical removal is the definitive treatment for most pheochromocytomas and paragangliomas. When appropriate, minimally invasive adrenalectomy is performed. For tumors outside the adrenal glands, specialized surgical approaches are used based on tumor location. Our experienced surgical teams prioritize precision, safety, and recovery.
Treatment for Metastatic or Recurrent Disease
When tumors recur or spread, additional options may include:
- MIBG radiotherapy
- Targeted therapies
- Chemotherapy
- Clinical trial participation
- Ongoing medical management of hormone-related symptoms
Prognosis & Follow-Up
With expert treatment and consistent follow-up, many patients achieve excellent long-term outcomes. Because PPGLs can recur—even years after treatment—lifelong monitoring is essential.
Follow-up care may include:
- Annual hormone testing
- Periodic imaging
- Blood pressure monitoring
- Genetic surveillance for hereditary conditions
Our endocrine specialists coordinate long-term care plans to detect recurrence early and support both patients and their families over time.
Why Choose Saint John’s for PPGL Care?
Patients choose Saint John’s for comprehensive, compassionate endocrine tumor care, including:
- Specialized expertise in adrenal and extra-adrenal neuroendocrine tumors
- High-volume experience in minimally invasive adrenal surgery
- Access to advanced imaging and genetic testing
- Multidisciplinary tumor board collaboration
- Personalized treatment and lifelong surveillance plans
Here at Saint john’s, care extends beyond diagnosis—our team partners with you through every stage of treatment and follow-up.
Frequently Asked Questions
Are pheochromocytomas cancerous?
Most pheochromocytomas and paragangliomas are benign, but a small percentage can be malignant. Lifelong follow-up is recommended for all patients.

Can pheochromocytoma or paraganglioma return after treatment?
Yes. Even after successful treatment, these tumors can recur years later. This is why lifelong follow-up—including periodic hormone testing, imaging, and blood pressure monitoring—is an essential part of ongoing care.
Should my family members be tested if I am diagnosed with pheochromocytoma or paraganglioma?
In some cases, yes. Because many pheochromocytomas and paragangliomas are linked to inherited genetic mutations, genetic counseling and testing may be recommended. Identifying a hereditary condition can help guide screening and early detection for family members.
If you have questions regarding Pheochromocytoma & Paraganglioma, or other endocrine conditions, please call today. Request an appointment



